
Research
The overarching theme of our research group is to use genetic approaches, both empirical and theoretical, to understand how evolutionary forces shaped the genetic architecture of complex traits within and between populations. To this end, we have been involved in a number of past and ongoing medical genetics studies in mapping genetic loci underlying human complex traits. We are also continually interested in investigating the evolutionary forces, namely demography and selection, that shaped the pattern of genetic variability and phenotypic distribution. We are particularly interested in global human populations with unique histories and ancestries, and our successes result from collaborating with innovative colleagues and thriving in resourceful consortiums.
The focus of the group mainly lies in the following areas:
Genetic basis of complex traits in unique global populations
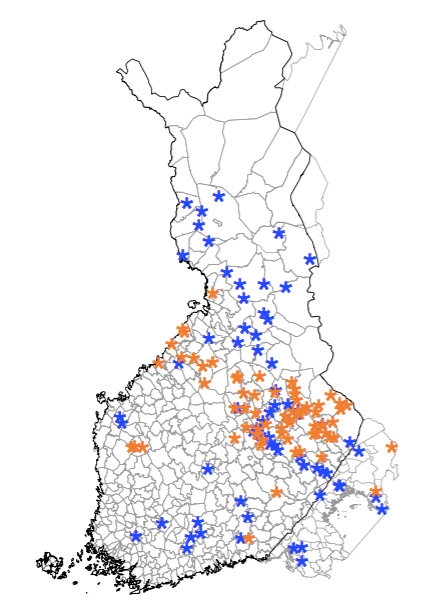
 Our group has led past and ongoing genetic studies in mapping genetic loci underlying human complex phenotypes such as anthropometric and cardiometabolic traits. Our focus has always been in studying unique populations with special demographic history. For example, we conducted one of the first GWAS of African-ancestry individuals for anthropometric traits (Hum. Mol. Genet. 2010). We have also leveraged the special population histories to map novel rare variant associated with height in Sardinians (Nat. Genet. 2015), as well as discovering scores of novel rare alleles associated with quantitative cardiometabolic traits in Northern and Eastern Finns (Nature 2019).
Our group has led past and ongoing genetic studies in mapping genetic loci underlying human complex phenotypes such as anthropometric and cardiometabolic traits. Our focus has always been in studying unique populations with special demographic history. For example, we conducted one of the first GWAS of African-ancestry individuals for anthropometric traits (Hum. Mol. Genet. 2010). We have also leveraged the special population histories to map novel rare variant associated with height in Sardinians (Nat. Genet. 2015), as well as discovering scores of novel rare alleles associated with quantitative cardiometabolic traits in Northern and Eastern Finns (Nature 2019).
We are currently focusing on understanding the genetic architecture of complex traits in understudied populations with unique histories and ancestries. There is a huge opportunity to leverage the evolutionary history of these populations to design and interpret genetic mapping studies (Front. Genet. 2021). However, we have shown that for a population like the Native Hawaiians, the lack of representation in genomic resources (e.g. a Polynesian haplotype reference panel for imputation) has been prohibitive to research (Hum. Mol. Genet. 2020). The lack of genomic resources and available cohort at scale contributed to the population not reaping the benefits of genomic medicine. For instance, polygenic score models based on the largest available GWAS perform more poorly among Native Hawaiians, and even worse among individuals enriched with Polynesian ancestry (Commun. Bio. 2025). In fact, the state-of-the-art imputation reference panel, TOPMed, still under-performs for many populations around the globe (AJHG 2024), including the Pacific Island populations. Therefore even though we know that epidemiologically Native Hawaiians have elevated risk for certain complex diseases, and we can show that this disease risks correlate with proportion of Polynesian genetic ancestry (PLoS Genet. 2021; but note that ancestry captures also non-genetic effects), we know very little of population-specific alleles that impacts disproportionately in these populations and thereby contributing to health disparities between populations. We are currently generating whole-genome sequencing data and constructing the genomic resources necessary to accelerate research in Polynesian populations, such as the recombination maps (Hum Genet 2024) and imputation reference panel. We are also forming a consortium of Pacific Island populations, including Native Hawaiians, Samoans, and other Pacific Islanders. In aggregate we will reach >13,000 Pacific Islanders with the goals of identifying more population-enriched variants such as CREBRF (Minster et al. Nat. Genet. 2016) across Pacific Islanders. We work hard to strengthen our interactions and engagement with the Native community, through interactions with the Native Hawaiian Community Advisory Board estsablished by the University of Hawaii Cancer Center, as well as through planned focus group to understand the community concerns with participating in genomic research.
In addition to Polynesian-ancestry or Pacific Island populations, we are also focusing on other diverse populations. For example, we have performed genetic mapping studies for Acute Lymphoblastic Leukemia (ALL) in multi-ethnic cohorts driven by Latinos (Leukemia 2022). We have also evaluated polygenic risk scores for ALL (HGG Adv 2023), investigated the impact of Indigenous American ancestry on ALL risk and its implication in the design and analysis of GWAS data (HGG Adv 2025). It is in this population that we have identified population-enriched risk alleles that appears to be under positive selection (Cell Genom 2024), which contributed to the elevated risk of ALL in Latino children. Finally, we are also taking an multiethnic approach to illuminate the complex genetic architecture of quantitative traits and diseases. For example, we were part of a multi-ethnic consortium that identified 71 novel loci associated with hematological traits that are not found in European ancestry populations (Cell 2020).
Fine-scale structure and demographic history of global populations

 One burgeoning movement in medicine is the incorporation of evolutionary principles. Medicine is classically concerned with only the most proximate causes of dysfunction and views diseases as defects of a once perfect machine. In contrast, evolutionary medicine strives to view diseases as the consequence of trade-offs and vulnerabilities due to interactions with our past environments. However, we need to first understand the demographic history and population structure of the population of interest, which in turns guide our design of the genetic studies to understand the genetic architecture and evolutionary courses of complex traits.
One burgeoning movement in medicine is the incorporation of evolutionary principles. Medicine is classically concerned with only the most proximate causes of dysfunction and views diseases as defects of a once perfect machine. In contrast, evolutionary medicine strives to view diseases as the consequence of trade-offs and vulnerabilities due to interactions with our past environments. However, we need to first understand the demographic history and population structure of the population of interest, which in turns guide our design of the genetic studies to understand the genetic architecture and evolutionary courses of complex traits.
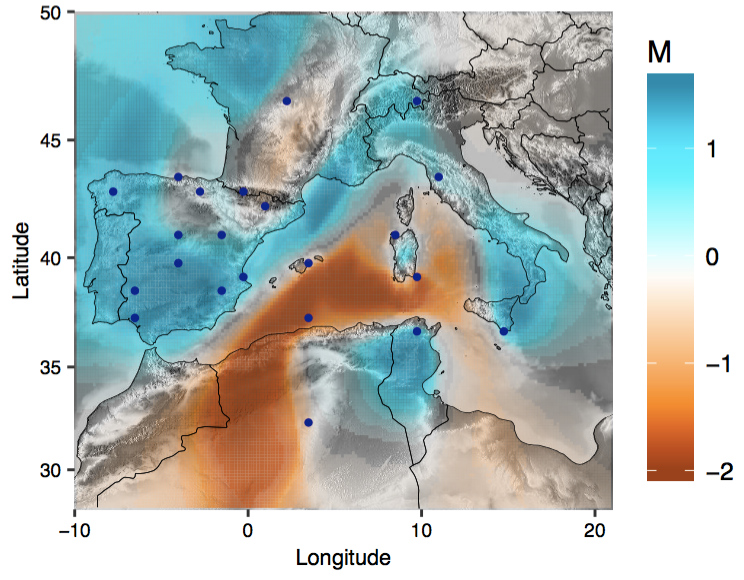
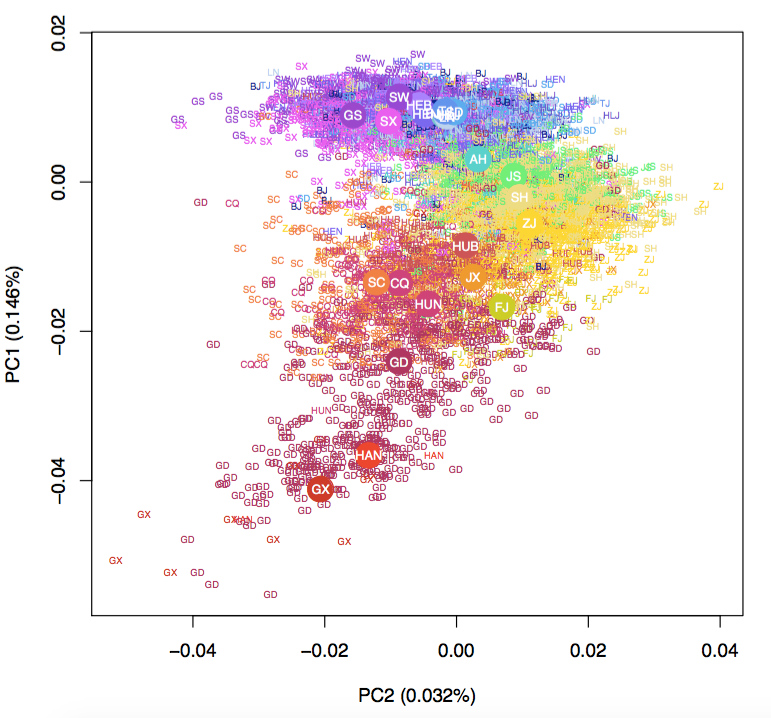 To this end, we have investigated the fine-scale structure and history of human populations known to have a unique past. We found geographical features that led to the genetic barrier and population structure in Sardinia (Nat. Genet. 2018). We conducted one of the first examinations of population structure in Han Chinese living in Taiwan (Hum. Mol. Genet. 2016), and identified additional geographical axis using whole genome sequence data in Han Chinese living in China (Mol. Biol. Evol. 2018). We characterized the fine-scale structure and the consequent enrichment of deleterious variation in Northern and Eastern Finland, thereby enabled unprecedented power to map rare variant associations (Nature 2019). Most recently, we also investigated the impact of demographic history and cultural practices on the pattern of genetic variation among Saudi Arabians (bioRxiv 2025). Future goals will continue to investigate global populations with unique histories or ancestries, such as the Native Hawaiians in the Multiethnic Cohort, or Han Chinese and Aboriginal Taiwanese in Taiwan Biobank. We will aim to understand in finer details of the population structure and admixture history of these populations to shed light on how differential susceptibility to diseases across populations may be rooted in their evolutionary histories.
To this end, we have investigated the fine-scale structure and history of human populations known to have a unique past. We found geographical features that led to the genetic barrier and population structure in Sardinia (Nat. Genet. 2018). We conducted one of the first examinations of population structure in Han Chinese living in Taiwan (Hum. Mol. Genet. 2016), and identified additional geographical axis using whole genome sequence data in Han Chinese living in China (Mol. Biol. Evol. 2018). We characterized the fine-scale structure and the consequent enrichment of deleterious variation in Northern and Eastern Finland, thereby enabled unprecedented power to map rare variant associations (Nature 2019). Most recently, we also investigated the impact of demographic history and cultural practices on the pattern of genetic variation among Saudi Arabians (bioRxiv 2025). Future goals will continue to investigate global populations with unique histories or ancestries, such as the Native Hawaiians in the Multiethnic Cohort, or Han Chinese and Aboriginal Taiwanese in Taiwan Biobank. We will aim to understand in finer details of the population structure and admixture history of these populations to shed light on how differential susceptibility to diseases across populations may be rooted in their evolutionary histories.
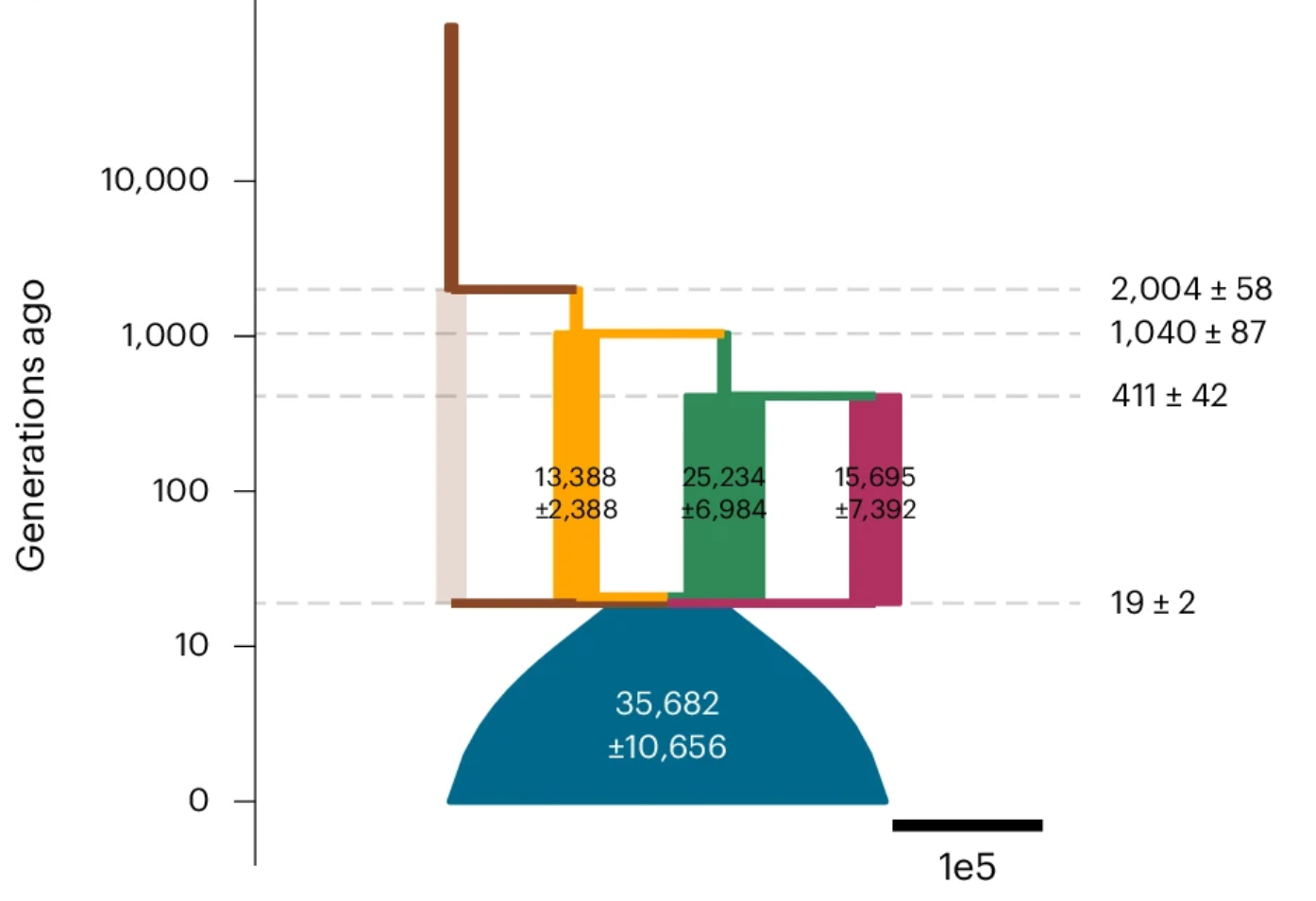 We are also developing innovative methods. Thanks to advancements that allows scalable inference of Ancestral Recombination Graphs genome-wide (Spiedel et al. Nat. Genet. 2018; Kelleher et al. Nat. Genet. 2018), we can approximate well the underlying genealogy that relates every one in the sample. Based on these sequences of genealogical trees, we proposed a novel metric of genetic relationship matrix (GRM) that computes the expected relatedness between pairs of individual given the genealogies. We found this "expected GRM" or eGRM is more sensitive to recent population structure, has the flexibility and potential to detect the varying nature of structure over time (AJHG 2022), and can be combined with local ancestry information to elucidate the ancestry-specific structure (AJHG 2025). We have subsequently used the local eGRM to improve the power of mapping quantitative trait loci, particularly in cases of allelic heterogeneity (AJHG 2023). Based on genealogical trees in the ARG, we have also devised methods that can estimate parameters of population demographic histories much more accurately than previous methods based on summary statistics, particularly for admixed populations (Nat. Genet. 2025).
We are also developing innovative methods. Thanks to advancements that allows scalable inference of Ancestral Recombination Graphs genome-wide (Spiedel et al. Nat. Genet. 2018; Kelleher et al. Nat. Genet. 2018), we can approximate well the underlying genealogy that relates every one in the sample. Based on these sequences of genealogical trees, we proposed a novel metric of genetic relationship matrix (GRM) that computes the expected relatedness between pairs of individual given the genealogies. We found this "expected GRM" or eGRM is more sensitive to recent population structure, has the flexibility and potential to detect the varying nature of structure over time (AJHG 2022), and can be combined with local ancestry information to elucidate the ancestry-specific structure (AJHG 2025). We have subsequently used the local eGRM to improve the power of mapping quantitative trait loci, particularly in cases of allelic heterogeneity (AJHG 2023). Based on genealogical trees in the ARG, we have also devised methods that can estimate parameters of population demographic histories much more accurately than previous methods based on summary statistics, particularly for admixed populations (Nat. Genet. 2025).
Natural Selection and Polygenic Adaptation
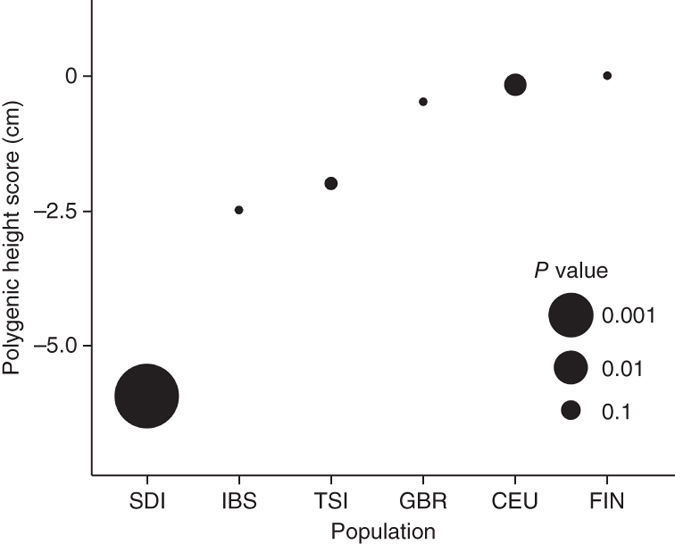 Demographic processes such as long-term isolation and bottleneck are not the only forces that shape the pattern of variation in our genome. Natural selection is another important factor that contributes to our genomic pattern of variation and distribution of complex traits. We have conducted genome-wide scans for signals of selection in human populations (Mol. Biol. Evol. 2018), as well as focusing on the polygenic signature of adaptation of copmlex traits, namely height. Using genome-wide summary statistics from GWAS we were the first to suggest that differentiation of adult human height across Europe is driven by polygenic adaptation (Nat. Genet. 2012). We later also demonstrated that the significant shorter stature in Sardinia is also driven by polygenic adaptation (Nat. Genet. 2015). Our study in human height stimulated the development of many methods aimed to detect such signatures in other complex traits as well as continued and ongoing discussion on interpreting polygenic scores from large scale GWAS (eLife 2019; also see Berg et al. eLife 2019). In light of the new insight that subtle uncorrected population structure could bias analysis of polygenic adaptation, we now leverage large-scale biobank data from distant, unrelated population such as the Biobank of Japan (BBJ) to unbiasly assess polygenic adaptation in Europeans. We found that differences in height between Sardinians and mainland Europeans are robustly attributed to polygenic adaptation, and we continue to find evidence consistent with natural selection driving the differences in height across mainland Europe, although the effect is weaker than previously believed (AJHG 2020). Using the same robust ascertainment approach, we also found that height-associated loci remain exceptionally differentiated across global populations, again supporting some form of natural selection that results in differentiation of height across populations (EJHG 2021)
Demographic processes such as long-term isolation and bottleneck are not the only forces that shape the pattern of variation in our genome. Natural selection is another important factor that contributes to our genomic pattern of variation and distribution of complex traits. We have conducted genome-wide scans for signals of selection in human populations (Mol. Biol. Evol. 2018), as well as focusing on the polygenic signature of adaptation of copmlex traits, namely height. Using genome-wide summary statistics from GWAS we were the first to suggest that differentiation of adult human height across Europe is driven by polygenic adaptation (Nat. Genet. 2012). We later also demonstrated that the significant shorter stature in Sardinia is also driven by polygenic adaptation (Nat. Genet. 2015). Our study in human height stimulated the development of many methods aimed to detect such signatures in other complex traits as well as continued and ongoing discussion on interpreting polygenic scores from large scale GWAS (eLife 2019; also see Berg et al. eLife 2019). In light of the new insight that subtle uncorrected population structure could bias analysis of polygenic adaptation, we now leverage large-scale biobank data from distant, unrelated population such as the Biobank of Japan (BBJ) to unbiasly assess polygenic adaptation in Europeans. We found that differences in height between Sardinians and mainland Europeans are robustly attributed to polygenic adaptation, and we continue to find evidence consistent with natural selection driving the differences in height across mainland Europe, although the effect is weaker than previously believed (AJHG 2020). Using the same robust ascertainment approach, we also found that height-associated loci remain exceptionally differentiated across global populations, again supporting some form of natural selection that results in differentiation of height across populations (EJHG 2021)
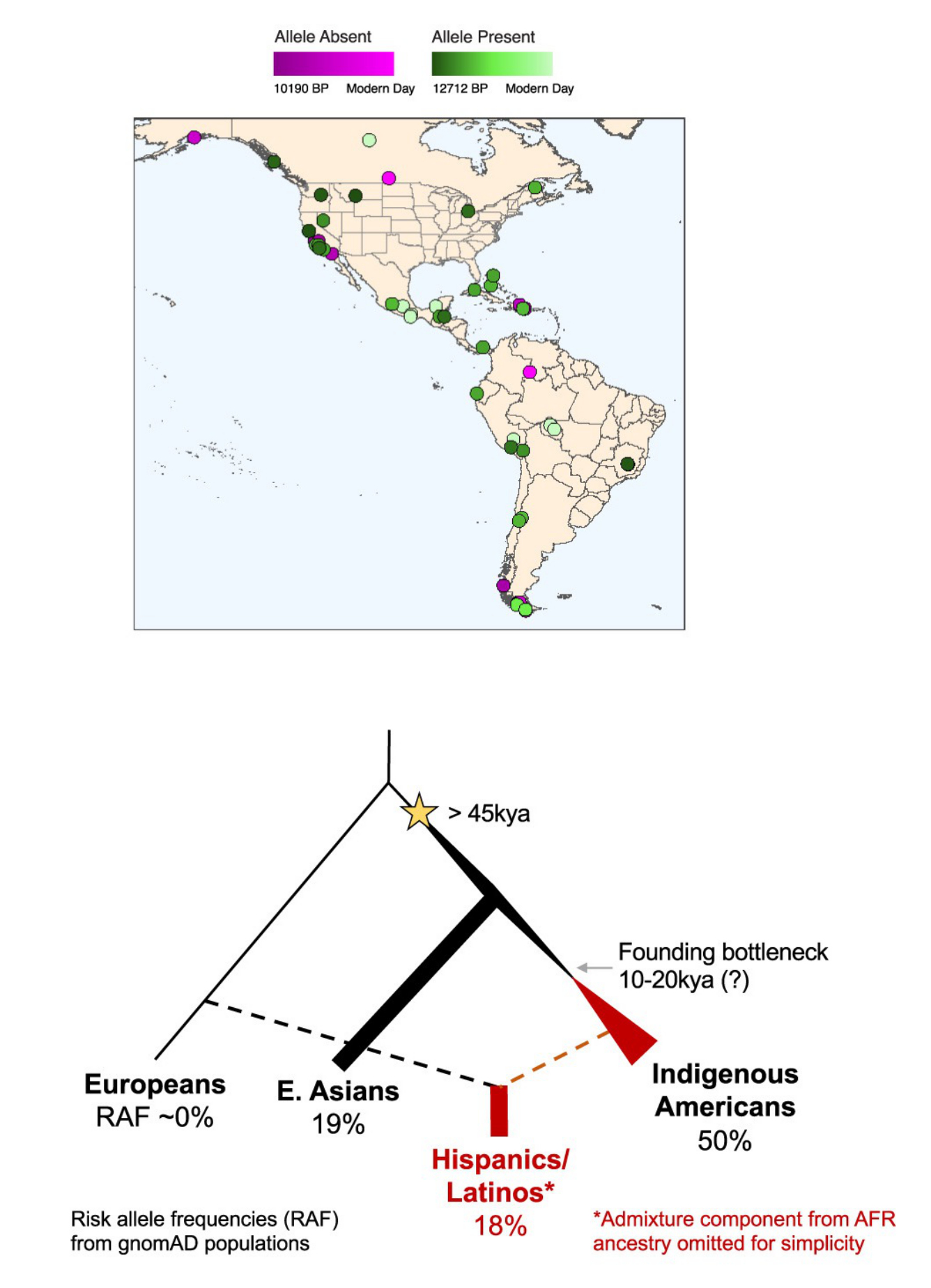 Moving forward, we will continue to investigate these imprints left in our genome by Natural Selection, and integrate external non-genetic information to contextualize these findings, which may reveal cases where increased susceptibility of certain traits may be a trade-off due to an evolutionary benefit in our past. For example, we have found that a population-enriched allele at IKZF1 confers elevated risk for childhood ALL and could be positively selected in the past in the Indigenous American populations (Cell Genom 2024). We will further evaluate more systematically if elevated risk of childhood leukemia found in Latino Americans is due to selection of alleles associated with, for instance, protection against infection in their past history. We will also examine the modern-day consequences to health due to potential past adaptation events in diverse human populations
Moving forward, we will continue to investigate these imprints left in our genome by Natural Selection, and integrate external non-genetic information to contextualize these findings, which may reveal cases where increased susceptibility of certain traits may be a trade-off due to an evolutionary benefit in our past. For example, we have found that a population-enriched allele at IKZF1 confers elevated risk for childhood ALL and could be positively selected in the past in the Indigenous American populations (Cell Genom 2024). We will further evaluate more systematically if elevated risk of childhood leukemia found in Latino Americans is due to selection of alleles associated with, for instance, protection against infection in their past history. We will also examine the modern-day consequences to health due to potential past adaptation events in diverse human populations
Datasets and Resources
Data Release (Chiang et al. Mol Bio Evol 2018)
- Allele frequencies stratified by provinces can be found at GGV (with google or twitter login, under "Converge"). link
Data Release (Cahoon et al. AJHG 2024)
- Disparity in imputation accuracies around the world: an interactive map summarizing the imputation efficacy of TOPMed reference panel for the plethora of global populations we assessed
Data Release (Dinh et al. Hum Genet 2024)
- Recombination maps for Native Hawaiian and Polynesian-Ancestry populations constructed using LDHat and IBDrecomb. github
Data Release (Malomane et al. bioRxiv 2025)
- Allele frequencies for 302 Saudi Arabian whole genome sequences. figshare
- Allele frequencies for array genotyped SNPs by inferred clusters among 3,352 Saudi Arabian individuals. figshare
Software Releases and Pipelines
expected Genetic Relationship Matrix (eGRM; Fan et al. AJHG 2022): constructs the expected GRM given inferred ancestral recombination graphs (ARG). github
triple-liftOver (Sheng et al. HGG Adv 2022): identifies variants mapping into a region inverted between genome builds to avoid downstream analysis problem such as imputation. github
gLike (Fan et al. Nat Genet 2025): estimate parameters of a demographic model given the genealogical trees. github
as-eGRM (Tang and Chiang, AJHG 2025): constructs the ancestry-specific eGRM with different weights given the ARG. github
Selected Publications
You can check pubmed or google scholar for the complete list of publication from the lab.
Group members are bolded; *, # denote equal contributions
- Langie J, Chan TF, Yang W, Kang AY, Morimoto L, Stram DO, Mancuso N, Ma X, Metayer C, Lupo PJ, Rabin KR, Scheurer ME, Wiemels JL, Yang JJ, de Smith AJ*, Chiang CWK*. “The impact of Indigenous American-like ancestry on risk of acute lymphoblastic leukemia in Hispanic/Latino children.” in press HGG Adv 2025. preprint.
- Tang J and Chiang CWK. “A genealogy-based approach for revealing ancestry-specific structures in admixed populations.” Am J Hum Genet. 2025 Aug 7;112(8):1906-1922. link
- Malomane DK, Williams MP, Huber CD, Mangul S*, Abedalthagafi M*, Chiang CWK*. “Patterns of population structure and genetic variation within the Saudi Arabian population.” bioRxiv 2025. preprint
- Lo YC, Tian H, Chan TF, Jeon S, Alatorre K, Dinh BL, Maskarinec G, Taparra K, Nakatsuka N, Yu M, Chen CY, Lin YF, Wilkens LR, Le Marchand L, Haiman CA, Chiang CWK. “Transferability of polygenic scores for anthropometric traits and Type II Diabetes to the Native Hawaiian population.” Commun Biol. 2025 Apr 23;8(1):651. link
- Fan C, Cahoon JL, Dinh BL, Ortega-Del Vecchyo VD, Huber CD, Edge MD, Mancuso N, Chiang CWK. “A likelihood-based framework for demographic inference from genealogical trees.” Nat Genet. 2025 Apr;57(4):865-874. link
- Cahoon JL, Rui X*, Tang E*, Simons C, Langie J, Chen M, Lo YC, Chiang CWK. “Imputation Accuracy Across Global Human Populations.” Am J Hum Genet. 2024 May 2;111. link
- de Smith AJ*, Wahlster L*, Jeon S*, Kachuri L, Black S, Cato LD, Nakatsuka N, Chan TF, Langie J, Mazumder S, Yang W, Gazal S, Eng C, Hu D, Buchard EG, Ziv E, Metayer C, Mancuso N, Yang JJ, Ma X, Wiemels JL, Yu F*, Chiang CWK*, Sankaran VG*. “A noncoding regulatory variant in IKZF1 increases acute lymphoblastic leukemia risk in Hispanic/Latino children.” Cell Genom. 2024 Apr 10;4(4):100526. link
- Dinh BL, Tang E, Taparra K, Nakatsuka N, Chen F, Chiang CWK. “Recombination map tailored to Native Hawaiians may improve robustness of genomic scans for positive selection.” Hum Genet. 2024 Jan;143(1):85-99. link
- Jeon S, Lo YC, Morimoto LM, Metayer C, Ma X, Wiemels JL, de Smith AJ, Chiang CWK. “Evaluating Genomic Polygenic Risk Scores for Childhood Acute Lymphoblastic Leukemia in Latinos.” HGG Adv. 2023 Sep 13:100239. doi: 10.1016/j.xhgg.2023.100239. link
- Link V, Schraiber JG, Fan C, Dinh B, Mancuso N, Chiang CWK, Edge MD. “Tree-based QTL mapping with expected local genetic relatedness matrices.” Am J Hum Genet. 2023 Dec 7;110(12):2077-2091. link
- Sheng X, Xia L, Cahoon JL, Conti DV, Haiman CA, Kachuri L, Chiang CWK. "Inverted genomic regions between reference genome builds in humans impact imputation accuracy and decrease the power of association testing." HGG Adv. 2022 Nov 11;4(1):100159. doi: 10.1016/j.xhgg.2022.100159. eCollection 2023 Jan 12. link
- Fan C, Mancuso N*, Chiang CWK*. “A genealogical estimate of genetic relationships.” Am J Hum Genet. 2022 Apr 6:S0002-9297(22)00112-4. doi: 10.1016/j.ajhg.2022.03.016. link
- Jeon S, de Smith AJ, Li S, Chen M, Chan TF, Muskens IS, Morimoto LM, Dewan AT, Mancuso N, Metayer C, Ma X, Wiemels JL, Chiang CWK. “Genome-wide trans-ethnic meta-analysis identifies novel susceptibility loci for childhood acute lymphoblastic leukemia.” Leukemia. 2022 Mar;36(3):865-868. doi: 10.1038/s41375-021-01465-1. Epub 2021 Nov 8. link
- Chiang CWK. “The opportunities and challenges of integrating population histories into genetic studies of global populations: a motivating example from Native Hawaiians.” Front Genet. 2021 Sep 27;12:643883. doi: 10.3389/fgene.2021.643883. link.
- Chen M and Chiang CWK. "Allele frequency differentiation at height-associated SNPs among contin-ental human populations.” Eur J Hum Genet. 2021 Oct;29(10):1542-1548. doi: 10.1038/s41431-021-00938-2. Epub 2021 Jul 15. link.
- Sun H*, Lin M*, Russell EM, Minster RL, Chan TF, Dinh BL, Naseri T, Reupena MS, Lum-Jones A, OLaGA Study Group, Cheng I, Wilkens LR, Le Marchand L, Haiman CA, Chiang CWK. “The impact of global and local Polynesian genetic ancestry on complex traits in Native Hawaiians.” PLoS Genet. 2021 Feb 11;17(2):e1009273. doi: 10.1371/journal.pgen.1009273. link.
- Chen MH, Raffield LM, Mousas A, Sakaue S, ..., Chiang CWK, Li B, Loos RJF, Astle WJ, Evangelou E, Sankaran VG, Okada Y, Soranzo N, Johnson AD, Reiner AP, Auer PL, Lettre G. "Trans-ethnic and ancestry-specific blood cell genetics in 746,667 individuals from 5 global populations." Cell. 2020 Sep 3;182(5):1198-1213.e14. link.
- Lin M, Caberto C, Wan P, Li Y, Lum-Jones A, Tiirikainen M, Pooler L, Nakamura B, Sheng X, Porcel J, Lim U, Setiawan VW, Le Marchand L, Wilkens LR, Haiman CA, Cheng I, Chiang CWK. “Population specific reference panels are crucial for the genetic analyses of Native Hawaiians: an example of the CREBRF locus.” Hum Mol Genet. 2020 Aug 3;29(13):2275-2284. doi: 10.1093/hmg/ddaa083. link.
- Chen M, Sidore C, Akiyama M, Ishigaki K, Kamatani Y, Schlessinger D, Cucca F, Okada Y, Chiang CWK. “Evidence of polygenic adaptation at height-associated loci in Sardinians using Biobank Japan.” Am J Hum Genet. 2020 Jul 2;107(1):60-71. doi: 10.1016/j.ajhg.2020.05.014. Epub 2020 Jun 12. link
- Locke AE*, Steinberg KM*, Chiang CWK*, Service S, Havulinna A, Stell L, Pirinen M, Abel HJ, Chiang CC, Fulton RS, Jackson AU, Kang CJ, Kanchi KL, Koboldt DC, Larson DE, Nelson J, Nicholas TJ, Pietila A, Ramensky V, Ray D, Scott LJ, Stringham HM, Vangipurapu J, Welch R, Yajnik P, Yin X, Eriksson JG, Ala-Korpela M, Jarvelin MR, Manniko M, Laivouri H, FinnGen Project, Dutcher SK, Stitziel NO, Wilson RK, Hall IM, Sabatti C, Palotie A, Salomaa V, Laakso M, Ripatti S, Boehnke M, Freimer NB. “Exome sequencing of Finnish isolates enhances rare-variant association power.” Nature. 2019 Aug;572(7769):323-328. doi: 10.1038/s41586-019-1457-z. Epub 2019 Jul 31. link
- Sohail M, Maier RM, Ganna A, Bloemendal A, Martin AR, Turchin MC, Chiang CWK, Hirschhorn J, Daly MJ, Patterson N, Neale B, Mathieson I, Reich D, Sunyaev SR. "Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies." Elife. 2019 Mar 21;8. pii: e39702. doi: 10.7554/eLife.39702. link
- Chiang CWK, Mangul S, Robles C, Sankararaman S. "A comprehensive map of genetic variation in the world's largest ethnic group - Han Chinese." Mol Bio Evol. 2018 Nov 1;35(11):2736-2750. doi: 10.1093/molbev/msy170. Epub 2018 Aug 30. link
- Chiang CWK, Marcus J, Sidore C,Al-asadi H, Biddanda A, Zoledziewska M, Pistis G, Steri M, Lohmueller K, Abecasis G, Schlessinger D, Cucca F, Novembre J. "Genomic History of the Sardinian Population" Nat Genet. 2018 Oct;50(10):1426-1434. Epub 2018 Sep 17. link
- Chen CH, Yang JH, Chiang CWK, Hsiung CN, Wu PE, Chang LC, Chang J, Song IW, Yang SL, Chen YT, Liu FT, Shen CY. "Population structure of Han Chinese in the modern Taiwanese population based on 10,000 participants in the Taiwan Biobank project." Hum Mol Genet. 2016 Dec 15;25(24):5321-5331.link
- Zoledziewska M*, Sidore C*, Chiang CWK*, Sanna S*, Mulas A, Steri M, Busonero F, Marcus JH, Marongiu M, Maschio A, Ortega Del Vecchyo VD, Floris M, Meloni A, Delitala A, Meloni A, Delitala A, Concas MP, Murgia F, Biino G, Vaccargiu S, Nagaraja R, Lohmueller KE; UK10K Consortium, Timpson NJ, Soranzo N, Tachmazidou I, Dedoussis G, Zeggini E; Understanding Society Scientific Group, Uzzau S, Jones C, Lyons R, Angius A, Abecasis GR#, Novembre J#, Schlessinger D#, Cucca F#. "Height-reducing variants and selection for short stature in Sardinia." Nat Genet. 2015 Nov; 47(11):1352-6. doi: 10.1038/ng.3403. Epub 2015 Sep 14. link
- Turchin MC*, Chiang CWK*, Palmer CD, Sankararaman S, Reich D, Genetic Investigation of ANthropometric Traits (GIANT) Consortium, Hirschhorn JN. "Evidence of widespread selection on standing variation in Europe at height-associated SNPs." Nat Genet. 2012 Sep;44(9):1015-9. doi: 10.1038/ng.2368. Epub 2012 Aug 19. link
- Chiang CWK, Liu CT, Lettre G, Lange LA, Jorgensen NW, Keating BJ, Vedantam S, Nock NL, Franceschini N, Reiner AP, Demerath EW, Boerwinkle E, Rotter JI, Wilson JG, North KE, Papanicolaou GJ, Cupples LA, Genetic Investigation of ANthropometric Traits (GIANT) consortium, Murabito JM, Hirschhorn JN. "Ultraconserved elements in the human genome: association and transmission analyses of highly constrained single-nucleotide polymorphisms." Genetics. 2012 Sep;192(1):253-66. Epub 2012 Jun 19. link
- Kang SJ*, Chiang CWK*, Palmer CD*, Tayo BO, Lettre G, Butler JL, Hackett R, Adeyemo AA, Guiducci C, berzins I, Nguyen TT, Feng T, Luke A, Shriner D, Ardlie K, Rotimi C, Wilks R, Forrester T, McKenzie CA, Lyon HN, Cooper RS, Zhu X#, Hirschhorn JN#. "Genome wide association of anthropometric traits in African and African derived populations." Hum Mol Genet. 2010 Jul 1;19(13):2725-38. Epub 2010 Apr 16. link